Scientific Problem | Overview
Why don't tissues in joints heal?
Previously proposed theories for the failure of the ACL to heal have included a deficient intrinsic cell response and vascular response.(2,4,7,14,19,22) Mechanical etiologies have also been proposed including lack of strength of the suture repair and fluid flow in and around the ligament and rupture site. Similar arguments have been made for meniscal healing(20) and rotator cuff suture repair.(15,23) Work directly addressing these hypotheses has met with limited in vivo success(6,21) and only marginal improvement in clinical outcomes in the past fifteen years.
The limited success seen with prior approaches prompted us to abandon the accepted doctrine for the failure of these tissues to heal and return to an unbiased failure analysis of the problem.
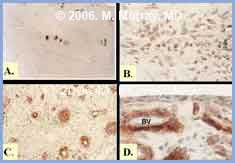 Fig 1. Histologic response of the human ACL to rupture
Fig 1. Histologic response of the human ACL to rupture
A. Histologic appearance of the normal ACL showing fibroblasts (blue nuclei; 40X).
B. Histologic appearance of ACL tissue 3 months after rupture showing increased cell density in the ligament ends (40X).
C. Area of increased capillary density in ACL ends 3 months after rupture (20X).
D. Synovial layer which has reformed over the ligament ends at 8 weeks after rupture (BV = blood vessel, 40X).
Sections are immunohistochemistry for alpha-smooth muscle actin (SMA) where red demonstrates a positive stain for SMA, with a blue counter-stain for cell nuclei.
To identify and define deficiencies in the healing process for these tissues, IRB approval was obtained and intact and injured human specimens of intra-articular tissues were collected over a 5 year period. Evaluated tissues included ligament, meniscus, cartilage and tendon.(1,8,10,13,16,17)
Examination and histomorphometric analyses were performed in a consistent manner for all tissues. Parameters of cell density, vascularity and extracellular matrix organization were systematically evaluated. Cellular functions including proliferative capability, migration characteristics and capacity for extracellular matrix production were evaluated for cells from normal(9,11) and injured tissues.(12,17)
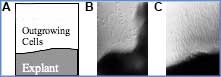 Fig 2. Proliferative & migratory capacity of cells in osteoarthritic articular cartilage in vitro. A) Schematic for orientation for B and C. B) Cell outgrowth at 7 days. C) Marked cell outgrowth after 14 days.
Fig 2. Proliferative & migratory capacity of cells in osteoarthritic articular cartilage in vitro. A) Schematic for orientation for B and C. B) Cell outgrowth at 7 days. C) Marked cell outgrowth after 14 days.
In comparing the tissues in the pre- and post-injury state, we identified an intrinsic cell and vascular response adjacent to the defect(10) (Fig 2B & 2C), as well as a remarkable capability for cells from these tissues to proliferate and migrate in vitro(9,12,17). Even the articular chondrocytes from patients with end-stage osteoarthritis had this capacity(17) (Fig. 3). However, at no time point was filling of the injury site observed (Fig. 3A & 3C). This was true for cartilage, meniscus and ACL.(8,10,13)
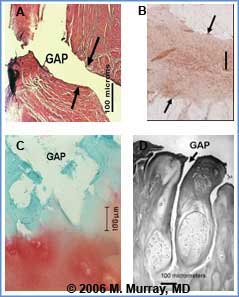 Fig. 3 Microscopic view of persistent wound site defects in ACL rupture after six weeks in vivo (A). Note the lack of filling of the defect. In contrast, the healing medial collateral ligament in (B) is completely filled in; even at only 7 days (Immunohistochemistry for TGF-b1 where red is a positive stain). The medial collateral ligament is located outside of the knee joint and is typically not exposed to synovial fluid after injury. Defects seen in the ACL are similar in appearance to those seen with chondral injury as early as 1 day after injury (C) where, again, the defects in articular cartilage do not fill in acutely or at later time points. The acute defects are likely the precursors to the clefts seen in osteoarthritic articular cartilage (D).
Fig. 3 Microscopic view of persistent wound site defects in ACL rupture after six weeks in vivo (A). Note the lack of filling of the defect. In contrast, the healing medial collateral ligament in (B) is completely filled in; even at only 7 days (Immunohistochemistry for TGF-b1 where red is a positive stain). The medial collateral ligament is located outside of the knee joint and is typically not exposed to synovial fluid after injury. Defects seen in the ACL are similar in appearance to those seen with chondral injury as early as 1 day after injury (C) where, again, the defects in articular cartilage do not fill in acutely or at later time points. The acute defects are likely the precursors to the clefts seen in osteoarthritic articular cartilage (D).
Lack of filling of the wound site for the tissues in joints was a key finding common to all tissues examined. In tissues outside of joints, the very first phase of the healing response is filling of the defect with a fibrin-platelet plug which bridges the wound edges(3). This plug, or scaffold, is then subsequently invaded by reparative cells that remodel the scaffold into healing fibrovascular scar. Formation of the early provisional scaffold is the first critical step in the wound healing process.
Inside the joint, bleeding occurs after injury, but no fibrin-platelet plug is observed to form, even at the injury site.(5) One possible explanation for this finding is that circulating intra-articular plasmin breaks down the fibrin plug as fast as it can form. Recent work has shown that after trauma to the joint, the production of plasmin is upregulated via the increased secretion of urokinase plasminogen activator(18)
With the additional circulating plasmin, the fibrin network is quickly destabilized within the joint environment and no fibrin-platelet plug forms. Premature loss of the fibrin-platelet plug would have the significant clinical benefit of preventing overall joint scarring and stiffness (arthrofibrosis) and thus maintenance of joint mobility after injury. However, this also requires the sacrifice of healing of the individual injured tissues, as the degradation of the fibrin-platelet plug in the overall joint also would remove it prematurely from any wound sites.
As formation of the fibrin-platelet plug is the essential first step for wound healing of musculoskeletal tissue outside the joint,(3) we believe that the loss of this fibrin-platelet plug inside the joint is a key mechanism behind the failure of tissues in the joint to heal (Fig. 5).
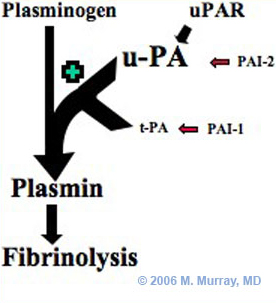 Fig 4. Proposed pathway for accelerated fibrinolysis after joint injury. Increased secretion of urokinase plasminogen activator (u-PA) results in high levels of plasmin in the inflammatory synovial fluid. This is likely the reason for the accelerated fibrinolysis noted in the joint after injury. (t-PA: tissue plasminogen activator. PAI-1 and 2: Plasminogen activator inhibitor 1 and 2. uPAR: urokinase-type plasminogen activator receptor)
Fig 4. Proposed pathway for accelerated fibrinolysis after joint injury. Increased secretion of urokinase plasminogen activator (u-PA) results in high levels of plasmin in the inflammatory synovial fluid. This is likely the reason for the accelerated fibrinolysis noted in the joint after injury. (t-PA: tissue plasminogen activator. PAI-1 and 2: Plasminogen activator inhibitor 1 and 2. uPAR: urokinase-type plasminogen activator receptor)
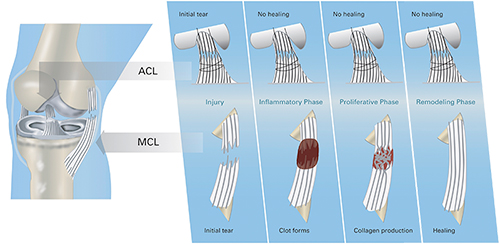 Fig. 5: Novel hypothesis of the failure of intra-articular healing. For the medial collateral ligament (MCL) which is outside the joint, injury is followed by formation of a provisional scaffold in the form of a fibrin clot. The scaffold is gradually remodeled as the tissue heals. No fibrin clot is found at the injury site of the ACL, which is inside the knee joint. Without a provisional scaffold, healing of the wound site cannot proceed.
Fig. 5: Novel hypothesis of the failure of intra-articular healing. For the medial collateral ligament (MCL) which is outside the joint, injury is followed by formation of a provisional scaffold in the form of a fibrin clot. The scaffold is gradually remodeled as the tissue heals. No fibrin clot is found at the injury site of the ACL, which is inside the knee joint. Without a provisional scaffold, healing of the wound site cannot proceed.
Our theory is that the premature failure of the provisional scaffold prevents healing of tissues inside joints (Fig. 5). This is an important change from previous mechanisms proposed for the individual tissues, which focused predominantly on the intrinsic cell and vascular responses.
Past research based on the cell-deficiency theory focused mainly on stimulation of cells in vitro with growth factors, or on cell transplantation into wound sites (including the use of stem cells and genetically modified cells).
Our preliminary work has shown that delivery of the appropriate bridge can stimulate the intrinsic healing response for tissues inside joints. This bridge can also be used as a foundation for the delivery of cytokine and cell-based treatments.
Stimulation tissue healing inside the joint could alter the way we approach these injuries. Enhanced primary suture repair and healing of these tissues would have several advantages over current treatments of resection and replacement. These procedures would have the advantage over simple resection of greater preservation of the joint tissues and thus potentially lower rates of osteoarthritis. Enhanced primary repair (Fig. 6) would also have advantages over current tissue replacement techniques, such as ACL reconstruction, including preservation of the complex attachment sites and innervation of these structures, thus retaining much of the biomechanical and proprioceptive function of these tissues as well as the gross anatomy.
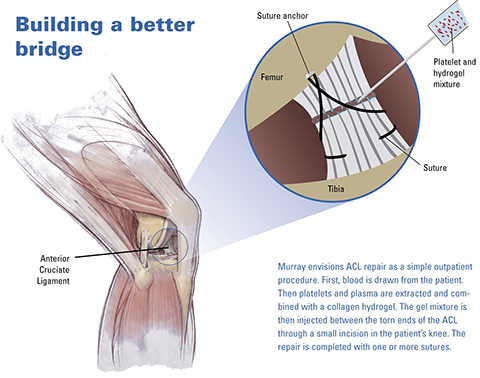 Fig. 6: Example of a potential clinical application. Stimulation of successful healing by enhancing suture repair with a biologic boost from a collagen-platelet hydrogel placed in the wound site could alter our approach toward these common injuries from resection and replacement toward repair and regeneration.
Fig. 6: Example of a potential clinical application. Stimulation of successful healing by enhancing suture repair with a biologic boost from a collagen-platelet hydrogel placed in the wound site could alter our approach toward these common injuries from resection and replacement toward repair and regeneration.