Our Research | Overview
The Gutierrez Laboratory is focused on unraveling the biologic basis for response and resistance to cancer therapies, and translating these findings into improved cancer therapies. Deep genetic characterization of human tumors over the past 5 decades has revolutionized our understanding of the genetic basis of cancer. However, our understanding of the biologic basis of treatment resistance lags far behind. Indeed, we still do not understand, in most cases, why patients with seemingly identical tumors can have strikingly different treatment responses. Additionally, the available therapeutic options we have for patients with such treatment-resistant tumors are too often woefully inadequate. The primary objectives of our work are to define mechanisms of treatment resistance, and devise novel effective therapies for these patients. Our approach typically begins with hypothesis-generating experiments using genetic or small molecule screens, coupled to functional genetics and biochemistry in genetically engineered and patient-derived models of human cancer.
This supports a model in which Wnt pathway activation impairs protein degradation, limiting cellular asparagine availability, and triggering a vulnerability to its enzymatic degradation using asparaginase.
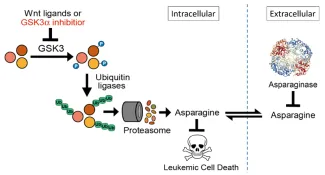
Pathobiology of asparaginase resistance
Asparaginase, an antileukemic enzyme that depletes asparagine, is a key therapeutic for acute lymphoblastic leukemias. However, resistance to asparaginase-based therapy has a poor prognosis, and its biologic basis is poorly understood. Using an unbiased genetic screen, we found that Wnt pathway activation induces profound asparaginase sensitivity in drug-resistant acute leukemias, but not in normal hematopoietic progenitors. Asparaginase sensitivity is caused by activation of a noncanonical (β-catenin) independent branch of Wnt signaling termed Wnt-dependent stabilization of proteins (Wnt/STOP), which inhibits GSK3-dependent protein ubiquitination and proteasomal degradation.
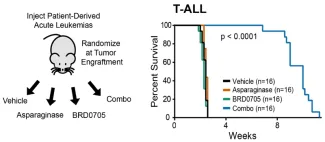
Wnt-induced asparaginase sensitivity is phenocopied by inhibition of GSK3α, and the combination of asparaginase and the GSK3α inhibitor BRD0705 has profound in vivo activity in patient-derived xenograft models of drug-resistant ALL.
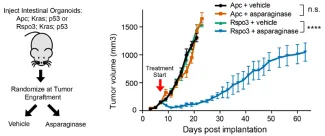
We then turned our attention to colorectal cancer, because this disease provides a unique context in which to test predictions from our model using disease-relevant mutations that can separate Wnt/β-catenin and Wnt-induced asparaginase sensitivity. Most (~85%) colorectal cancers have mutations (such as APC loss or β-catenin activation) that we predicted would activate β-catenin without inhibiting GSK3 or triggering asparaginase sensitivity. However, a minority of colorectal cancers are driven by mutations (such as R-spondin fusions) that stimulate Wnt-induced inhibition of GSK3, which we predicted would trigger asparaginase sensitivity.
We found that asparaginase monotherapy lacked activity against APC or β-catenin mutant colorectal cancer, but was profoundly toxic to R-spondin fusion tumors.
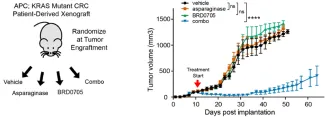
Additionally, pharmacologic inhibition of GSK3α was sufficient to trigger asparaginase sensitivity in APC and β-catenin mutant CRC, providing a potential therapeutic approach for most patients with colorectal cancer.
Our ongoing studies suggest that GSK3-dependent protein degradation represents a previously unrecognized cellular response to amino acid starvation. Our interests in this area are to decipher the molecular regulation of this adaptive response to amino acid starvation, and to define the molecular determinants of response and resistance to the combination of GSK3α inhibition and asparaginase. We are also working with the group of our collaborator Flo Wagner at Broad Institute to solve the pharmacologic constraints of existing GSK3α inhibitors, which we hope will yield small molecule GSK3α inhibitors suitable for human clinical trials.
Genetic screens for chemotherapy resistance
Ongoing studies in the lab are focused on leveraging unbiased genetic screens to unravel the biology of resistance to potentially curative cancer therapies. These studies have implicated a number of unexpected enzymes and metabolic pathways as determinants of response to cancer therapeutics, which represent the focus of ongoing studies in the lab. A recurrent theme emerging from these studies is that tumors can develop chemotherapy resistance via mechanisms that are non-overlapping with those that allow normal cells to tolerate these drugs, providing ideal targets for therapeutic intervention. We are eager to test how far this concept can revolutionize cancer therapeutics.