2021
February 27, 2021
Dr. Kulandaisamy A, a bioinformatics PhD from the Indian Institute of Technology Madras, Dr. Yihan Zuo, a cardiology PhD from the Macau University of Science and Technology, and Dr. Rongbin Zheng, a bioinformatics PhD from the Tongji University and a former visiting student at Harvard will join the lab as postdocs.
February 27, 2021
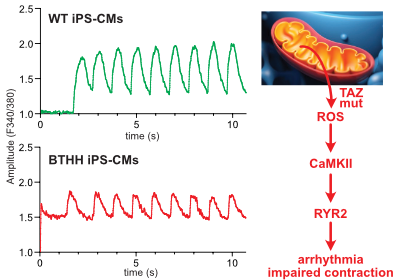
Liu, X. et al., Circulation in press
Barth syndrome cardiomyocytes from human iPSCs and mice have abnormal calcium handling that is partially due to activation of CaMKII. Barth syndrome mice had increased vulnerability to arrhythmia, perhaps reflecting the abnormal calcium handling.
January 20, 2021
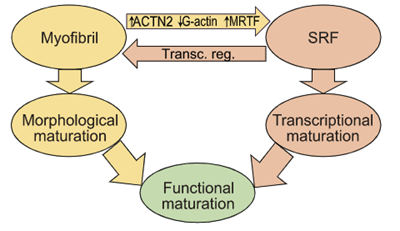
Guo, Cao et. al. PNAS - Read the article
Sarcomere assembly promotes MRTF-SRF signaling to drive a feed-forward circuit that stimulates cardiomyocyte maturation.
2020
August 25, 2020
CAT-D opening is scheduled for 9/10/20.
March 10, 2020
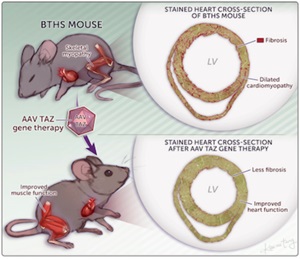
Wang, S et al., Circ. Res. In press. Read the article
AAV gene therapy improved survival and heart and skeletal muscle function in a new mouse model of Barth syndrome.
March 5, 2020
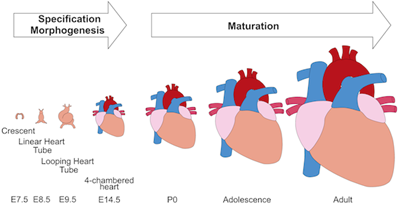
In press in Circulation Research.
Cardiomyocyte maturation is the process whereby proliferative and glycolytic fetal cardiomyocytes become specialized to forcefully and efficiently contract billions of times in an animal’s lifespan. This review article by Yuxuan Guo and William Pu summarizes our current understanding of mechanisms that promote and coordinate the process of cardiomyocyte maturation.
2019
October 29, 2019
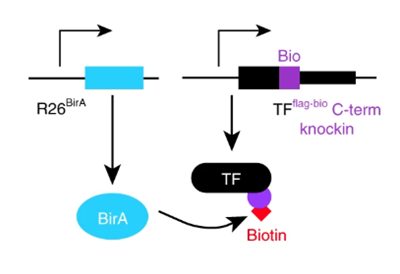
Mapping transcription factors (TFs) occupancy is essential for understanding transcriptional programs. Here Akerberg, Gu, and colleagues use biotinylated knockin alleles of key cardiac TFs (GATA4, NKX2-5, MEF2A, MEF2C, SRF, TBX5, TEAD1) to map their genome-wide occupancy in the fetal and adult mouse heart, providing insight into the cardiac transcriptional regulatory network.
July 21, 2019
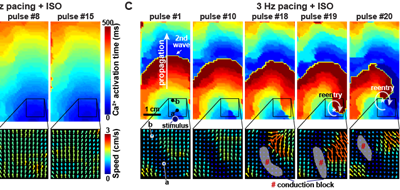
In an equal collaboration between the Pu and Parker labs, Park, Zhang, et al. describe development of an engineered human tissue model of CPVT. Using this model, the investigators dissected the molecular pathway linking exercise to unmasking of the arrhythmia mutation. The study revealed that CaMKII phosphorylation of RYR2-S2814 is essential for expression of multiple RYR2 mutations.
Background:
Modeling of human arrhythmias using induced pluripotent stem cell-derived cardiomyocytes has focused on single cell phenotypes. However, arrhythmias are the emergent properties of cells assembled into tissues, and the impact of inherited arrhythmia mutations on tissue-level properties of human heart tissue has not been reported.
Methods:
Here, we report an optogenetically-based, human engineered tissue model of catecholaminergic polymorphic ventricular tachycardia (CPVT), an inherited arrhythmia caused by mutation of the cardiac ryanodine channel (RYR2) and triggered by exercise. We developed a hiPSC-CM-based platform to study the tissue-level properties of engineered human myocardium. We investigated pathogenic mechanisms in CPVT, by combining this novel platform with genome editing.
Results:
In our model, CPVT tissues were vulnerable to develop reentrant rhythms when stimulated by rapid pacing and catecholamine, recapitulating hallmark features of the disease. These conditions elevated diastolic Ca2+ levels and increased temporal and spatial dispersion of Ca2+ wave speed, creating a vulnerable arrhythmia substrate. Using Cas9 genome editing, we pinpointed a single catecholamine-driven phosphorylation event, RYR2-S2814 phosphorylation by Ca2+-calmodulin-dependent protein kinase II (CaMKII), that is required to unmask the arrhythmic potential of CPVT tissues.
Conclusions:
Our study illuminates the molecular and cellular pathogenesis of CPVT and reveals a critical role of CaMKII-dependent reentry in the tissue-scale mechanism of this disease. We anticipate that this approach will be useful to model other inherited and acquired cardiac arrhythmias.
June 23, 2019
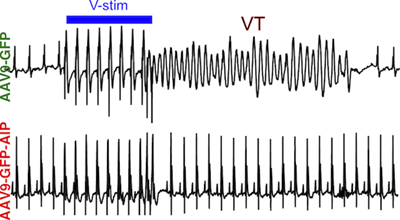
Background:
Catecholaminergic polymorphic ventricular tachycardia (CPVT), an inherited cardiac arrhythmia characterized by adrenergically triggered arrhythmias, is inadequately treated by current standard of care. Ca2+/calmodulin-dependent protein kinase II (CaMKII), an adrenergically activated kinase that contributes to arrhythmogenesis in heart disease models, is a candidate therapeutic target in CPVT. However, translation of CaMKII inhibition has been limited by the need for selective CaMKII inhibition in cardiomyocytes. Here we tested the hypothesis that CaMKII inhibition using a cardiomyocyte-targeted gene therapy strategy would suppress arrhythmia in CPVT mouse models.
Methods:
We developed AAV9-GFP-AIP, an adeno-associated viral vector in which a potent CaMKII inhibitory peptide (AIP), is fused to GFP and expressed from a cardiomyocyte selective promoter. The vector was delivered systemically. Arrhythmia burden was evaluated using invasive electrophysiology testing in adult mice. AIP was also tested on induced pluripotent stem cells (iPSC) derived from CPVT patients with different disease-causing mutations to determine the effectiveness of our proposed therapy on human iPSC-derived cardiomyocytes (iPSC-CMs) and different pathogenic genotypes.
Results:
AAV9-GFP-AIP was robustly expressed in the heart without significant expression in extra-cardiac tissues, including the brain. Administration of AAV9-GFP-AIP to neonatal mice with a known CPVT mutation ( RYR2R176Q/+) effectively suppressed ventricular arrhythmias induced by either β-adrenergic stimulation or programmed ventricular pacing, without significant proarrhythmic effect. Intravascular delivery of AAV9-GFP-AIP to adolescent mice transduced ~50% of cardiomyocytes and was effective in suppressing arrhythmia in CPVT mice. iPSC-CMs derived from two different CPVT patients with different pathogenic mutations demonstrated increased frequency of abnormal calcium release events, which was suppressed by a cell-permeable form of AIP.
Conclusions:
This proof-of-concept study showed that AAV-mediated delivery of a CaMKII peptide inhibitor to the heart was effective in suppressing arrhythmias in a murine model of CPVT. CaMKII inhibition also reversed the arrhythmia phenotype in human CPVT iPSC-CMs models with different pathogenic mutations.