Ryanodine Receptor 1 Related Myopathies (RYR1-RM) are a group of congenital muscle diseases related and characterized by the involvement of a mutation in the ryanodine receptor calcium channel embedded in the membrane of the sarcoplasmic reticulum (SR). RYR1-RMs are the most common type of nondystrophic muscle diseases, which include Malignant Hyperthermia (MH), Congenital Fiber Type Disproportion (CFTD), Centronuclear Myopathy (CMD), Multiminicore Disease (MmD), and Central Core Disease (CCD) (Amburgey et al. 2011; Amburgey et al. 2013; Jungbluth et al. 2018; Snoeck et al. 2015; Treves et al. 2008).
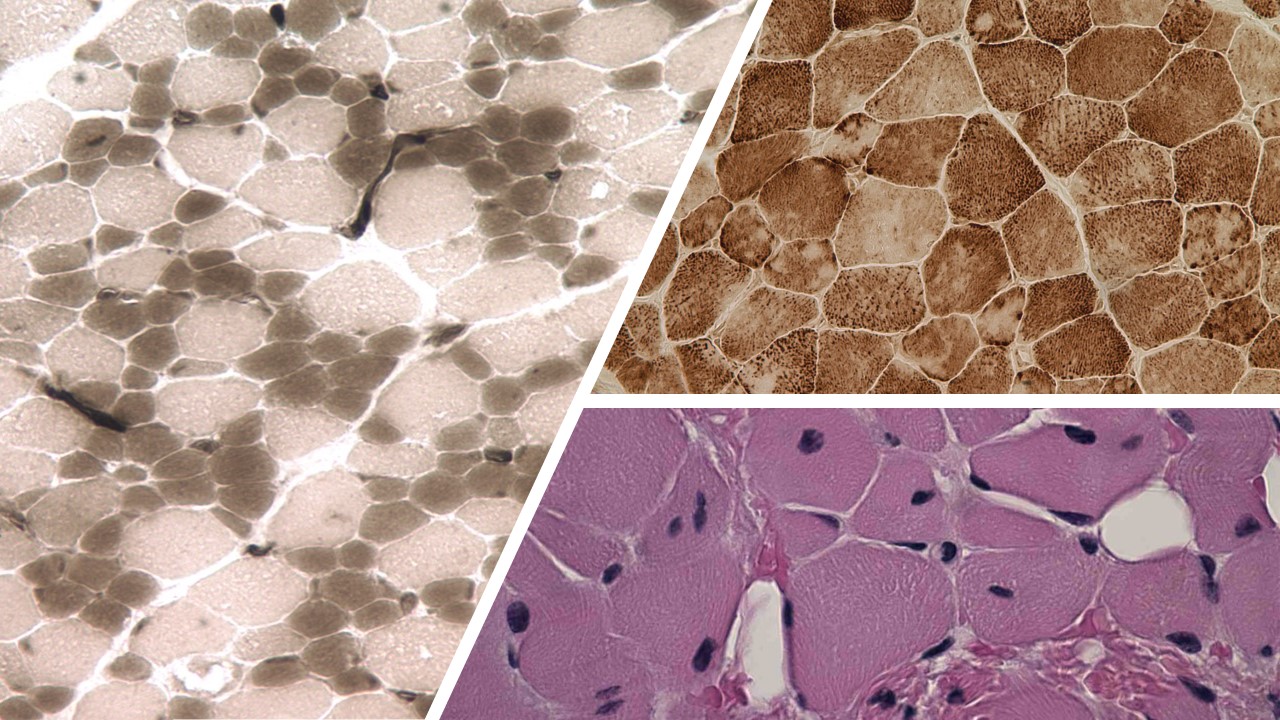
Left to right; top to bottom: CFTD, MMD, and CMD.
The RYR1 channel is anchored directly within the SR membrane in skeletal muscle, and is composed of four identical protein subunits that form a mushroom-like shape with the broad end exposed to the cytosol and the narrow ends residing within the SR. RYR1 is primarily tasked with connecting motor neuron signaling to muscle contraction in a process known as excitation-contraction coupling (ECC). A membrane potential travels down to a voltage sensor known as the dihydropyridine receptor (DHPR), which undergoes a conformational change that will mechanically interact with the RYR1 structure and open it, allowing the large stores of calcium held within the SR (between 1000X to 10,000X the concentration of the rest of the cell interior) out into the cytosol to initiate muscle contraction (Fill and Copello 2002; Franzini-Armstrong and Jorgenson 1994; Lanner et al. 2010; Rebbeck et al. 2011; Treves et. al. 2017). There are two other isoforms of the ryanodine receptor gene within the human genome, RYR2 and RYR3, which both provide similar functions pertaining to calcium release and signaling. However, RYR1 is the only isoform that plays a predominant role in ECC (Nakai et al. 1997).
Genetics and pathology
With more than 700 identified gene variants, it is difficult to parse out the exact dynamics of how and to what extent mutations of the RYR1 gene play (Litman et al. 2018). Clinical cases of patients with RYR1-RM encompass a large swath of symptoms including skeletal abnormalities, proximal muscle weakness, respiratory muscle weakness, extraocular muscle weakness, and dysmorphic cranial features. Upon biopsy, muscle fiber samples show atypical proportions of different muscle type fibers, centrally located nuclei, and muscle fibers with cores formed within them (North et al. 2014; Sato et al. 2008; Snoeck et al. 2015). RYR1 mutations can be inherited via both dominant and recessive means. Although there are also reported cases of adult-onset RYR1-RM diseases, most symptoms usually manifest during early childhood development (Jungbluth et al. 2009; Loseth et al. 2013).
Research
A wide variety of preclinical models have been used to study the function and etiological significance of RYR1, due to the central nature of its role in muscle development and activity. Predominantly, these have been cell culture or rodent models, but also include drosophila, porcine, c. elegans, canine, avian, equine, and zebrafish models (Lawal et al. 2020). Recently there has been progress constructing various mouse models that mimic the phenotype of certain RYR1-RM seen in patients, a potentially exciting development that bears future investigation (Brennan et al. 2019; Elbaz et al. 2019).
In particular our lab studies a spontaneously produced mutation of RYR1 in zebrafish that eliminates expression of the RYR1b gene. Although on a genetic level this is not the same as in humans, the phenotype resulting in the larval zebrafish musculature is remarkably akin to attributes of MmD observed in human skeletal muscle and presents a good opportunity to study the development of the disease (Hirata et al. 2007). Zebrafish provide a number of advantages as a model of myopathies, as they develop quickly and are encased within an embryonic membrane that allows for the diffusion of the surrounding water in and out. This yields a unique opportunity to expose the growing larvae to potential therapeutic substances in early stages of development when myofibrillar organization is not yet complete (Chagovetz et al. 2019; Maves 2014; Wu et al. 2011). The zebrafish myotome is very well ordered, and unlike mammalian skeletal muscle, the slow and fast twitch muscle fibers are segregated from each other, which is of particular interest when studying the varying effects of RYR1 (Stickney et al. 2000). By using this model, we hope to better understand the mechanisms involved in RYR1 activity and discover applicable treatments to ameliorate symptoms of RYR1-RM in patients with congenital myopathies.
References
Amburgey, K., N. McNamara, L.R. Bennett, M.E. McCormick, G. Acsadi, and J.J. Dowling. “Prevalence of congenital myopathies in a representative pediatric United States population.” Ann Neurol. 70 no. 4 (2011): 662–5.
Amburgey, K., A. Bailey, J.H. Hwang, M.A. Tarnopolsky, C.G. Bonnemann, L. Medne, K.D. Mathews, J. Collins, J.R. Daube, G.P. Wellman et al. “Genotype-phenotype correlations in recessive RYR1-related myopathies.” Orphanet J. Rare Dis. 8 (2013): 117. doi:10.1186/1750-1172-8-117.
Brennan, S., M. Garcia-Castaneda, A. Michelucci, N. Sabha, S. Malik, L. Groom, L. Wei-La-Pierre, J.J. Dowling, and R.T. Dirksen. “Mouse model of severe recessive RYR1-related myopathy.” Hum. Mol. Genet. 28, no. 18 (2019): 3024–3036 doi:10.1093/hmg/ ddz105.
Chagovetz, A.A., D.K. Shaw, E. Ritchie, K. Hoshijima and D.J. Grunwald. “Interactions among ryanodine receptor isotypes contribute to muscle fiber type development and function.” Disease Models & Mechanisms 13 (2020): dmm038844. Doi:10.1242/dmm.038844.
Elbaz, M., A. Ruiz, C. Bachmann, J. Eckhardt, P. Pelczar, E. Venturi, C. Lindsay, A.D. Wilson, A. Alhussni, T. Humberstone et al. “Quantitative RyR1 reduction and loss of calcium sensitivity of RyR1Q1970fsX16+A4329D cause cores and loss of muscle strength.” Hum. Mol. Genet. 28, no. 18 (2019): 2987–2999 doi:10.1093/hmg/ ddz092.
Fill, M. and J.A. Copello. “Ryanodine receptor calcium release channels.” Physiol. Rev. 82 (2002): 893-922 doi:10.1152/physrev.00013.2002.
Franzini-Armstrong, C., and A.O. Jorgensen. “Structure and development of E-C coupling units in skeletal muscle.” Annu Rev Physiol 56 (1994): 509–534.
Hirata, H., T.Watanabe, J. Hatakeyama, S.M. Sprague, L. Saint-Amant, A. Nagashima, W. W. Cui, W. Zhou, and J. Y. Kuwada. “Zebrafish relatively relaxed mutants have a ryanodine receptor defect, show slow swimming and provide a model of multi-minicore disease.”Development 13, no. 4 (2007): 2771-2781 doi:10.1242/dev.004531.
Jungbluth, H., S. Lillis, H. Zhou, S. Abbs, C. Sewry, M. Swash, F. Muntoni. “Late- onset axial myopathy with cores due to a novel heterozygous dominant mutation in the skeletal muscle ryanodine receptor (RYR1) gene.” Neuromuscul Disord. 19, no. 5 (2009): 344–7.
Jungbluth, H., S. Treves, F. Zorzato, A. Sarkozy, J. Ochala, C. Sewry, R. Phadke, M. Gautel, and F. Muntoni. “Congenital myopathies: disorders of excitation-contraction coupling and muscle contraction.” Nat. Rev. Neurol. 14 (2018): 151-167. Doi:10.1038/nrneurol.2017.191.
Lanner, J.T., D.K. Georgiou, A.D. Joshi, and S.L. Hamilton. “Ryanodine receptors: structure, expression, molecular details, and function in calcium release.” Cold Spring Harb. Perspect. Biol. 2, a003996 (2010). Doi:10.1101/cshperspect. A003996.
Lawal, T.A., E.S. Wires, N. L. Terry, J.J. Dowling and J.J. Todd. “Preclinical model systems of ryanodine receptor 1-related myopathies and malignant hyperthermia: a comprehensive scoping review of works published 1990– 2019.” Orphanet Journal of Rare Diseases 15 (2020): 113 https://doi.org/10.1186/s13023-020-01384-x.
Litman, R.S., S.M. Griggs, J.J. Dowling, S. Riazi. “Malignant hyperthermia susceptibility and related diseases.” Anesthesiology 128, no. 1 (2018): 159–67.
Loseth, S., N.C. Voermans, T. Torbergsen, S. Lillis, C. Jonsrud, S. Lindal, E.J. Kamsteeg, M. Lammens, M. Broman, G. Dekomien et al. “A novel late-onset axial myopathy associated with mutations in the skeletal muscle ryanodine receptor (RYR1) gene.” J Neurol. 260, no. 6 (2013): 1504–10.
Maves, L. “Recent advances using zebrafish animal models for muscle disease drug discovery.” Expert Opin Drug Discovery 9, no. 9 (2014): 1033–45.
Nakai, J., T. Ogura, F. Protasi, C. Franzini-Armstrong, P.D. Allen, K.G. Beam. “Functional nonequality of the cardiac and skeletal ryanodine receptors.” Proc Natl Acad Sci USA 94 (1997): 1019–1022.
North, K.N., C.H. Wang, N. Clarke, H. Jungbluth, M. Vainzof, J.J. Dowling, K. Amburgey, S. Quijano-Roy, A.H. Beggs, C. Sewry et al. “Approach to the diagnosis of congenital myopathies.” Neuromuscul Disord. 24 no. 2(2014):97–116.
Rebbeck, R.T., Y. Karunasekara, E.M. Gallant, P.G. Board, N.A. Beard, M.G. Casarotto, A.F. Dulhunty. “The β(1a) subunit of the skeletal DHPR binds to skeletal RyR1 and activates the channel via its 35-residue C-terminal tail.” Biophys J. 100, no. 4 (2011):922–30.
Sato, I., S. Wu, M. C. A. Ibarra, Y. K. Hayashi, H. Fujita, M. Tojo, S. J. Oh, I. Nonaka, S. Noguchi, and I. Nishino. “Congenital neuromuscular disease with uniform type 1 fiber and RYR1 mutation.” Neurology 70 (2008): 2.
Snoeck, M., B.G.M. van Engelen, B. Küsters, M. Lammens, R. Meijer, J.P.F. Molenaar, J. Raaphorst, C.C. Verschuuren-Bemelmans, C.S.M. Straathof, L.T.L. Sie et al. “RYR1-related myopathies: a wide spectrum of phenotypes throughout life.” Eur J Neurol. 22, no. 7 (2015): 1094–112.
Stickney, H.L., M.J.F. Barresi, and S.H. Devoto. “Somite development in zebrafish.” Dev. Dyn. 219 (2000): 287-303. doi:10.1002/1097-0177(2000)9999:9999<::AID-DVDY1065>3.0.CO;2-A.
Treves, S., H. Jungbluth, F. Muntoni, F. Zorzato. “Congenital muscle disorders with cores: the ryanodine receptor calcium channel paradigm.” Curr Opin Pharmacol 8 (2008): 319–326.
Treves, S., H. Jungbluth, N. Voermans, F. Muntoni, and F. Zorzato. “Ca(2+) handling abnormalities in early-onset muscle diseases: Novel concepts and perspectives.” Semin. Cell Dev. Biol. 64 (2017): 201-212. Doi:10.1016/j.semcdb. 2016.07.017.
Wu, H.H.T., C. Brennan, and R. Ashworth. “Ryanodine receptors, a family of intracellular calcium ion channels, are expressed throughout early vertebrate development.” BMC Res. Notes 4 (2011) 541. doi:10.1186/1756-0500- 4-541.
This page was last updated November 27, 2020.