Multiminicore disease mainly occurs when there are mutations in either the SELENON or RYR1 genes and consists of a group of congenital neuromuscular diseases that are clinically and genetically heterogenous. Patients present with non-progressive weakness with symptoms varying from mild to severe and life threatening. Most cases (75%) of mutiliminicore disease tend to exhibit the ‘classic’ form with core and neck muscles weakness beginning early in life which eventually result in the spine becoming rigid, and in some cases lead to scoliosis (Beggs and Agrawal 1993; Zorzato et al. 2007). As the stiffening occurs the respiratory system also begins to be affected and often requires some form of nocturnal ventilation (Caggiano et al. 2017). The initial onset of symptoms in this classic form often results in a delay in the motor milestones in affected children; however most affected individuals do gain and maintain the ability to walk for the majority of their lives (Jungbluth 2007; Ferreiro et al. 2002). The atypical form of multimincore tends to be milder, and without the respiratory complications; whereas the moderate and antenatal forms tend to affect the joints with either loosing or stiffening respectively (Beggs and Agrawal 1993). Other medical problems that have been also observed in affected individuals include heart disease, and increased risk for malignant high blood temperature with anesthesia have been associated with mutations in the RYR1 gene (Monnier et al. 2003)); whereas mutations in the SELENON gene have primarily resulted in respiratory insufficiency (Guis et al. 2004).
Multiminicore disease is diagnosed by examining a small piece of a muscle biopsy under a microscope. Patients’ muscle sections stained for mitochondria activity (NADH staining) show multiple small areas that are not stained which are referred to as core lesions or multiminicores. The core lesions correspond to areas of the muscle depleted in the mitochondria and are the hallmark of the multiminicore myopathy (Zorzato et al. 2007; Jungbluth 2007; Ferreiro et al. 2002).
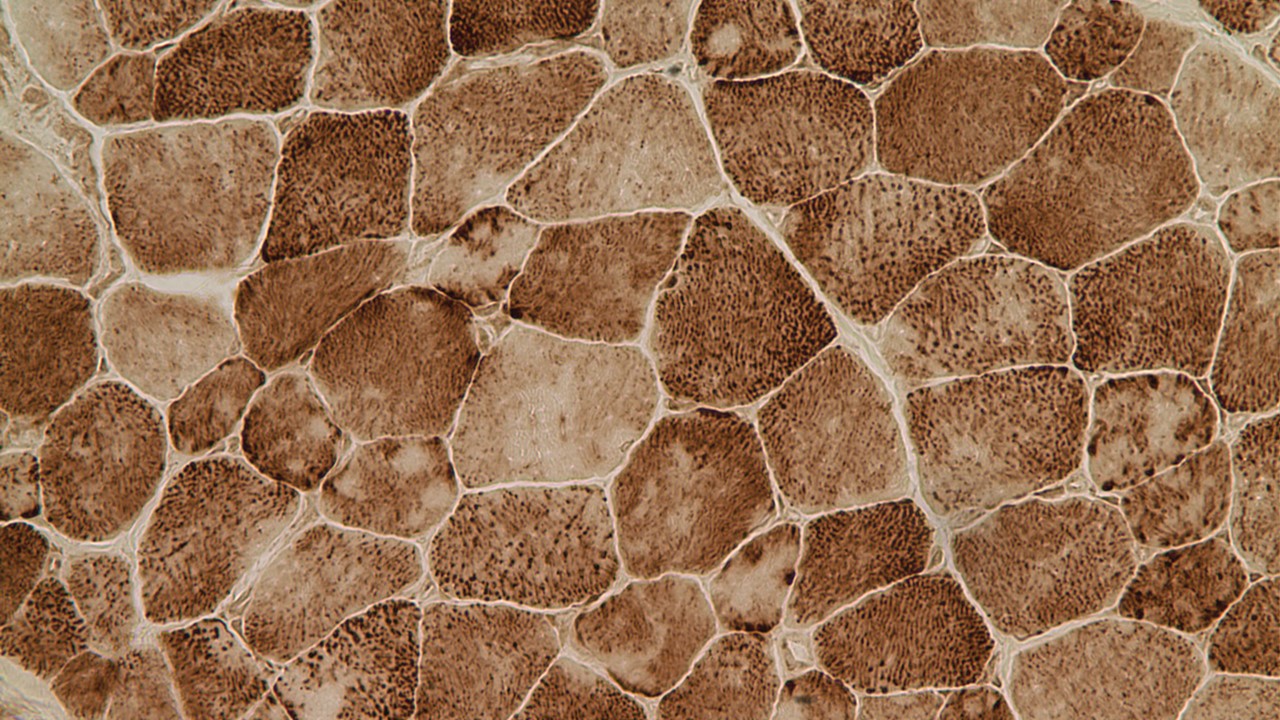
Muscle cells exhibiting the classic minicores (lighter core areas within the cells) observed in Multiminicore disease.
Genetics
Multiminicore disease appears to be caused by alterations in certain genes that are involved in muscle function and/or development which are inherited in an autosomal recessive form (Zorzato et al. 2007). To date mutations have been found in two genes, Selenoprotein N1 (SELENON; formerly SEPN1; Ferreiro et al. 2002) and Ryanodine receptor 1 (RYR1; Monnier et al. 2003) which together account for about half the cases. A third of all multiminicore disease cases are due to mutations of the SELENON gene (Moghadaszadeh et al. 2001; Zorzato et al. 2007). In addition, SELENON mutations cause a phenotypically distinct condition called Rigid Spine Muscular Dystrophy (Moghadaszadeh et al. 2001; Ferreiro et al. 2002). Rigid Spine Muscular Dystrophy and multiminicore disease as a result of SELENON mutations have recently been grouped together as a condition now referred to as SELENON - Related Myopathies (Varone et al. 2019). The function of selenoprotein N is important for calcium homeostasis through redox regulation of SERCA channel which controls the calcium flux from cytosol to the endo/sarcoplasmic reticulum. Ryanodine receptor on the other hand, is a calcium channel that releases calcium from the sarcoplasmic reticulum to the cytosol upon stimulation of the muscle (Treves et al. 2005; Varone et al. 2019).
Research
One of the goals of the research done at the Beggs laboratory is to further understand genes and proteins involved in multiminicore disease. Understanding the genetic cause(s) of congenital myopathies will increase our knowledge of pathophysiological pathways involved in these diseases and provide insights into future development of specific treatments and therapies. We hope that our findings will be useful for improving diagnosis and treatment of these conditions. If you are the parent of a child with this condition, if you yourself are affected, or if you are the healthcare provider of a person with multiminicore disease, you may be able to help us expand our knowledge in this condition and perhaps develop better tests, treatments and therapies.
References
Beggs, A. H. and P. B. Agrawal. "Multiminicore Disease." In Genereviews((R)), edited by M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, and A. Amemiya. Seattle (WA): University of Washington, Seattle. University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved., 1993.
Ben Hamida, M., F. Hentati, and C. Ben Hamida. "[Multiminicore Disease in a Rigid Spine Syndrome]." Rev Neurol (Paris) 143, no. 4 (1987): 284-9.
Caggiano, S., S. Khirani, I. Dabaj, E. Cavassa, A. Amaddeo, J. O. Arroyo, I. Desguerre, P. Richard, R. Cutrera, A. Ferreiro, B. Estournet, S. Quijano-Roy, and B. Fauroux. "Diaphragmatic Dysfunction in Sepn1-Related Myopathy." Neuromuscul Disord 27, no. 8 (2017): 747-55.
Ferreiro, A., S. Quijano-Roy, C. Pichereau, B. Moghadaszadeh, N. Goemans, C. Bonnemann, H. Jungbluth, V. Straub, M. Villanova, J. P. Leroy, N. B. Romero, J. J. Martin, F. Muntoni, T. Voit, B. Estournet, P. Richard, M. Fardeau, and P. Guicheney. "Mutations of the Selenoprotein N Gene, Which Is Implicated in Rigid Spine Muscular Dystrophy, Cause the Classical Phenotype of Multiminicore Disease: Reassessing the Nosology of Early-Onset Myopathies." Am J Hum Genet 71, no. 4 (2002): 739-49.
Guis, S., D. Figarella-Branger, N. Monnier, D. Bendahan, G. Kozak-Ribbens, J. P. Mattei, J. Lunardi, P. J. Cozzone, and J. F. Pellissier. "Multiminicore Disease in a Family Susceptible to Malignant Hyperthermia: Histology, in Vitro Contracture Tests, and Genetic Characterization." Arch Neurol 61, no. 1 (2004): 106-13.
Jungbluth, H. "Multi-Minicore Disease." Orphanet J Rare Dis 2 (2007): 31.
Moghadaszadeh, B., N. Petit, C. Jaillard, M. Brockington, S. Quijano Roy, L. Merlini, N. Romero, B. Estournet, I. Desguerre, D. Chaigne, F. Muntoni, H. Topaloglu, and P. Guicheney. "Mutations in Sepn1 Cause Congenital Muscular Dystrophy with Spinal Rigidity and Restrictive Respiratory Syndrome." Nat Genet 29, no. 1 (2001): 17-8.
Monnier, N., A. Ferreiro, I. Marty, A. Labarre-Vila, P. Mezin, and J. Lunardi. "A Homozygous Splicing Mutation Causing a Depletion of Skeletal Muscle Ryr1 Is Associated with Multi-Minicore Disease Congenital Myopathy with Ophthalmoplegia." Hum Mol Genet 12, no. 10 (2003): 1171-8.
Treves, S., A. A. Anderson, S. Ducreux, A. Divet, C. Bleunven, C. Grasso, S. Paesante, and F. Zorzato. "Ryanodine Receptor 1 Mutations, Dysregulation of Calcium Homeostasis and Neuromuscular Disorders." Neuromuscul Disord 15, no. 9-10 (2005): 577-87.
Varone, E., D. Pozzer, S. Di Modica, A. Chernorudskiy, L. Nogara, M. Baraldo, M. Cinquanta, S. Fumagalli, R. N. Villar-Quiles, M. G. De Simoni, B. Blaauw, A. Ferreiro, and E. Zito. "Selenon (Sepn1) Protects Skeletal Muscle from Saturated Fatty Acid-Induced Er Stress and Insulin Resistance." Redox Biol 24 (2019): 101176.
Zorzato, F., H. Jungbluth, H. Zhou, F. Muntoni, and S. Treves. "Functional Effects of Mutations Identified in Patients with Multiminicore Disease." IUBMB Life 59, no. 1 (2007): 14-20.
This page was last updated October 23, 2020.